ATTR amyloidosis occurs when the transthyretin protein, normally stable and helpful for transporting substances in the blood, becomes unstable and forms harmful amyloid deposits. This can affect the heart (leading to cardiomyopathy), nerves, and other areas. It typically starts subtly in later life, but hereditary forms may begin earlier. While not fully curable, early intervention can manage symptoms and slow decline.
For more details, see resources like the Amyloidosis Research Consortium at https://www.arci.org.
There are two main types:
- wild-type (non-hereditary, linked to aging)
- hereditary (genetic mutations), with debates on prevalence varying by region and ethnicity
It seems likely that hereditary forms may present earlier and involve more systems.
Treatments focus on stabilizing TTR or reducing its production, with drugs like tafamidis showing benefits in slowing progression; however, controversy exists around access and efficacy in advanced stages, emphasizing the need for personalized care.
The condition arises from TTR protein misfolding, either due to natural aging (wild-type) or inherited gene changes (hereditary). Factors like male gender, older age, and certain ethnic backgrounds (e.g., African ancestry for specific mutations) increase risk. While the precise “why” involves protein instability, lifestyle and environmental influences are not well-established triggers.
Diagnosis usually involves blood tests, imaging like echocardiograms or bone scans, and sometimes biopsies to confirm amyloid type. Treatments include medications to stabilize TTR (e.g., tafamidis) or silence its production (e.g., patisiran), plus supportive care for heart and nerve symptoms. Emerging options show promise, but consult specialists for tailored plans. Lifestyle adjustments, like a heart-healthy diet and gentle exercise, may help manage symptoms.
Transthyretin (ATTR) amyloidosis: overview
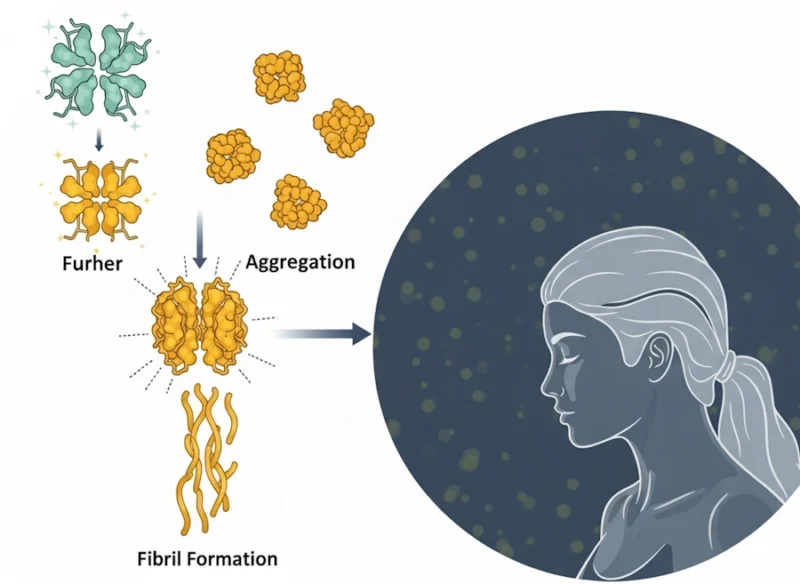
Transthyretin amyloidosis (ATTR amyloidosis) is a multisystemic, progressive disorder characterized by the extracellular deposition of misfolded transthyretin (TTR) protein as amyloid fibrils, leading to organ dysfunction. TTR, a 55-kDa homotetrameric protein primarily synthesized in the liver, functions physiologically as a transporter for thyroxine (T4) and retinol-binding protein (RBP).
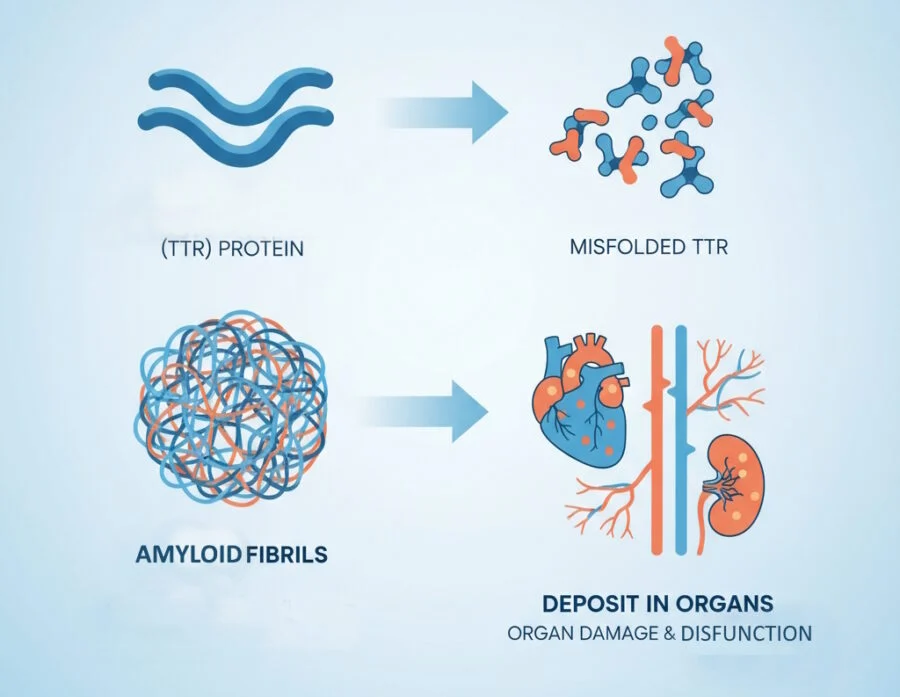
However, under certain conditions, TTR dissociates into monomers that misfold, aggregate, and form insoluble amyloid fibrils. This process disrupts tissue architecture and function, predominantly affecting the heart, peripheral nerves, and autonomic nervous system.
ATTR amyloidosis exists in two primary subtypes:
- wild-type ATTR (ATTRwt),
- age-related condition without genetic mutations, and variant ATTR (ATTRv), a hereditary form driven by autosomal dominant TTR gene mutations.
Although once considered rare, improved diagnostic tools have revealed ATTR amyloidosis as an underrecognized cause of heart failure, neuropathy, and other age-associated morbidities, with significant implications for clinical management.
Epidemiology and prevalence
Epidemiological data on ATTR amyloidosis indicate a higher burden than previously estimated, with heterogeneity across regions, demographics, and subtypes.
A systematic literature review synthesizing global evidence reported ATTR cardiomyopathy (ATTR-CM) prevalence ranging from 6.1 per million in the United States to 232 per million in Portugal, with incidence rates increasing over time (e.g., from 5.0 to 18.0 per million person-years in France between 2011 and 2017).
ATTRwt predominates in older populations, with prevalence estimates of 54.9 per million among those aged 65 years or older in the US, and up to 100 per million person-years in elderly Japanese cohorts.
In high-risk groups, such as patients with heart failure with preserved ejection fraction (HFpEF), ATTR-CM prevalence reaches approximately 13%, while in those undergoing transcatheter aortic valve replacement (TAVR) for severe aortic stenosis, it is around 16%.
Up to 40% prevalence has been noted in Afro-Caribbean individuals with heart failure and increased ventricular wall thickness.
ATTRv exhibits regional endemicity, with the p.V50M mutation common in Portugal, Sweden, and Japan, and p.V142I prevalent in individuals of African ancestry (3%-4% carrier rate).
Mixed phenotypes (e.g., cardiomyopathy and polyneuropathy) occur in about 33.5% of cases, as per the Transthyretin Amyloidosis Outcomes Survey (THAOS) registry.
Mortality data show 2-year risks of 10%-30% for ATTRwt-CM and 10%-52% for ATTRv-CM, with median survival varying from 12 to 80 months.
The disease burden is amplified by diagnostic delays (often 3-5 years), socioeconomic disparities, and limited data from underrepresented regions like Africa and South America, where endemic areas (e.g., Brazil, Argentina) suggest underreporting.
Projections anticipate rising incidence with aging populations and enhanced screening, underscoring the need for global registries to refine estimates and address gaps in ATTR polyneuropathy (ATTR-PN) data.
Causes and pathophysiology: how and why it starts
The pathogenesis of ATTR amyloidosis centers on TTR tetramer instability, dissociation into monomers, partial unfolding, and subsequent aggregation into amyloid fibrils. In ATTRwt, this instability is age-associated, with mechanisms incompletely understood but linked to oxidative stress, proteostatic decline, and environmental factors that promote misfolding in individuals over 60, predominantly males (85%-90%).
No genetic mutation is involved, yet amyloid deposition preferentially affects the heart, leading to restrictive cardiomyopathy.
In ATTRv, over 130 pathogenic TTR variants (e.g., p.V142I, p.V50M, p.T80A) destabilize the tetramer via altered protein conformation, inherited autosomally dominantly with incomplete penetrance.
The p.V142I variant, common in Afro-Caribbeans (23% of US ATTR-CM cases), exemplifies ethnic predisposition.
Disease onset varies: early (third to fifth decade) in endemic areas like Portugal for p.V50M, often with neuropathy, versus later (after 50) for cardiac-dominant phenotypes.
Fibril deposition causes direct toxicity—prefibrillar oligomers impair cardiomyocyte contractility and relaxation—while mature fibrils mechanically disrupt tissue, inducing inflammation and fibrosis.
Extracardiac involvement includes nerves (sensorimotor and autonomic neuropathy), eyes (vitreous opacities, glaucoma), kidneys (proteinuria, renal failure), and musculoskeletal system (carpal tunnel syndrome, spinal stenosis).
The “why” involves a interplay of genetic predisposition, aging, and potentially modifiable factors like inflammation, though exact triggers remain elusive, with research suggesting sequence-dependent denaturation energetics as a key determinant.
Symptoms and what people experience
People with ATTR amyloidosis often report increasing fatigue, shortness of breath, swelling in the legs, irregular heartbeats, and numbness or pain in the hands and feet.
Condition can mimic common aging issues, leading to delayed diagnosis.
Some experience carpal tunnel syndrome or digestive problems years before heart issues appear. It’s important to discuss persistent symptoms with a doctor, as they can significantly impact daily life but vary widely among individuals.
Vascular symptoms
Clinical manifestations of ATTR amyloidosis are insidious, often mimicking common conditions like hypertension or idiopathic neuropathy, contributing to diagnostic delays. Cardiac symptoms dominate in ATTRwt and many ATTRv cases, presenting as progressive HFpEF with dyspnea on exertion, orthopnea, fatigue, peripheral edema, and low-output states in advanced stages.
Arrhythmias, such as atrial fibrillation (15%-70% prevalence, higher in ATTRwt), conduction blocks, syncope, and stroke, are frequent due to amyloid infiltration. Echocardiographic features include increased ventricular wall thickness (>12 mm), biatrial enlargement, and reduced global longitudinal strain with apical sparing.
Neurological symptoms
Neurologic symptoms, more prominent in ATTRv (e.g., p.V50M), include peripheral sensorimotor neuropathy starting distally (paresthesia, hypesthesia, pain in feet), progressing proximally to motor deficits (foot drop, weakness). Autonomic dysfunction manifests as orthostatic hypotension, gastrointestinal disturbances (diarrhea, constipation), impotence, urinary incontinence, and dry mouth.
Extracardiac signs like bilateral carpal tunnel syndrome (often preceding cardiac symptoms by 5-10 years), lumbar spinal stenosis, biceps tendon rupture, and trigger finger serve as red flags.
Patient experiences vary by subtype and stage: early ATTRv may involve subtle neuropathy impacting mobility and quality of life, while ATTRwt often presents with debilitating heart failure in later life, leading to frequent hospitalizations. Mixed phenotypes combine cardiac and neurologic burdens, with plasma neurofilament light chain levels correlating with neuropathy severity.
Symptoms may evolve over 10-20 years, with death from cardiac failure, infection, or renal complications if untreated.
| Symptom Category | Common Manifestations | Typical Onset and Progression | Affected Subtypes |
|---|---|---|---|
| Cardiac | Dyspnea, edema, fatigue, arrhythmias (e.g., AF), syncope | Late (after 60 in ATTRwt; variable in ATTRv) | Both, predominant in ATTRwt |
| Neurologic (Peripheral) | Numbness, paresthesia, pain, weakness in extremities | Early (30-50s in ATTRv) | Primarily ATTRv |
| Autonomic | Orthostatic hypotension, GI issues, impotence, incontinence | Variable, often with neuropathy | ATTRv > ATTRwt |
| Musculoskeletal | Carpal tunnel syndrome, spinal stenosis, tendon rupture | Precedes cardiac by 5-10 years | Both, common in ATTRwt |
| Other (Ocular/Renal) | Vitreous opacities, glaucoma, proteinuria | Rare to moderate | ATTRv variants |
Diagnosis
Accurate diagnosis requires a high index of suspicion, especially in elderly males with unexplained HFpEF or neuropathy.
Noninvasive imaging has revolutionized detection: technetium-99m pyrophosphate (99mTc-PYP) scintigraphy offers >99% sensitivity and 87%-100% specificity for ATTR-CM (Perugini grade 2-3 uptake, excluding monoclonal proteins via serum/urine immunofixation and free light chain assay).
Echocardiography reveals hypertrophic patterns with strain abnormalities, while cardiac MRI shows late gadolinium enhancement and elevated extracellular volume (sensitivity 86%-93%).
Biomarkers like NT-proBNP and troponin are elevated in symptomatic disease but less useful for screening; lower circulating TTR and RBP4 levels indicate amyloidogenesis.
For confirmation, tissue biopsy (e.g., abdominal fat pad, sensitivity 60%-80% for ATTR) with Congo red staining and mass spectrometry differentiates ATTR from AL amyloidosis.
Genetic testing identifies TTR variants in ATTRv, recommended for family screening starting 10 years before expected onset. Challenges include distinguishing from hypertensive cardiomyopathy and avoiding overreliance on imaging without ruling out light-chain amyloidosis.
Treatment and management
Therapeutic paradigms have evolved rapidly, focusing on TTR stabilization, silencing, or fibril disruption, alongside supportive care.
- TTR stabilizers like tafamidis (80 mg daily, FDA-approved 2019) reduce mortality (29.5% vs. 42.9% placebo at 30 months, ATTR-ACT trial) and hospitalizations, with long-term data confirming benefits in both subtypes.
- Acoramidis (phase 3, >90% stabilization) showed superior outcomes in hierarchical endpoints (win ratio 1.8).
- Diflunisal (250 mg twice daily) offers off-label stabilization but requires monitoring for renal/gastrointestinal risks.
- Gene silencers target hepatic TTR production: patisiran (IV siRNA, APOLLO trial) improves neuropathy and cardiac parameters; vutrisiran (subcutaneous, HELIOS-B trial) reduces mortality by 36% and enhances function.
- Inotersen and eplontersen (antisense oligonucleotides) show efficacy but need thrombocytopenia/glomerulonephritis monitoring.
- Newly developing therapies include CRISPR-Cas9 (NTLA-2001) for gene editing, monoclonal antibodies (NI006, NNC6019) for amyloid clearance, and disruptors like doxycycline/TUDCA.
- Supportive management addresses heart failure (diuretics, avoiding digoxin/calcium blockers), arrhythmias (pacemakers), and neuropathy (pain medications like duloxetine).
- Liver/heart transplantation is reserved for select ATTRv cases.
- Lifestyle recommendations include low-sodium diets, moderate exercise (e.g., walking, yoga), weight management, and avoiding extremes in temperature due to autonomic risks. Multidisciplinary care involving cardiologists, neurologists, and genetic counselors is essential.
| Treatment Class | Examples | Mechanism | Key Trial Outcomes | Approvals and Considerations |
|---|---|---|---|---|
| TTR Stabilizers | Tafamidis, Acoramidis, Diflunisal | Bind to TTR tetramer, prevent dissociation | Mortality reduction (30%-43%), improved function | FDA-approved (tafamidis); monitor renal function |
| Gene Silencers | Patisiran, Vutrisiran, Inotersen, Eplontersen | RNA interference or antisense to reduce TTR production | Reduced events (28%-36%), better neuropathy/QoL | Approved for neuropathy; ongoing for CM; infusion reactions, thrombocytopenia risks |
| Fibril Disruptors | Doxycycline/TUDCA, NI006, NTLA-2001 | Degrade deposits or edit TTR gene | Reduced amyloid load (phase 1-2); halted progression | Investigational; tolerability issues in some |
| Supportive | Diuretics, Pacemakers, Pain Meds | Symptom management | N/A | Standard for HF/neuropathy; avoid certain cardiac drugs |
Prognosis and directions
Prognosis depends on subtype, stage, and treatment: untreated ATTR-CM median survival is 3-5 years from diagnosis, improved to >5 years with stabilizers. Early intervention in ATTRv carriers via screening enhances outcomes.
Future research emphasizes precision medicine, with trials like CARDIO-TTRansform evaluating eplontersen and ATTRibute-CM for acoramidis. Challenges include high costs (e.g., tafamidis) and access in low-resource settings, but advancements in biomarkers and immunotherapies promise earlier detection and reversal of deposits.